COVID Vaccine Design
Categories: COVID Publications
Designing a COVID Vaccine with the Gifford Lab at MIT CSAIL
For posterity’s sake I am sharing a Tweet thread for the paper I was involved with Computationally Optimized SARS-CoV-2 MHC Class I and II Vaccine Formulations Predicted to Target Human Haplotype Distributions:
These tweets are for the publication of the paper in Cell Systems and for the bioRxiv preprint:
Our SARS-CoV-2 vaccine design has been published in Cell Systems! Re-written for clarity and with additional results, including what peptides can augment existing S protein vaccines to significantly boost their population coverage.
— Trenton Bricken (@TrentonBricken) July 28, 2020
Link to paper: https://t.co/QXBTNhrZXy https://t.co/QnLhxGpEQY pic.twitter.com/U65n6FGdD9
In case you would rather read the full text rather than the Tweet thread it is below. This Tweet thread is with respect to the preprint, the full paper is here. The results remained unchanged but sections were re-written for clarity and some extra analysis performed:
Proud to have contributed to designing a SARS-CoV-2 vaccine! Our vaccine significantly outperforms previously published SARS-CoV-2 vaccine designs in maximizing population coverage (which we calculate using our intuitive yet novel population coverage objective) and quite likely in the peptides actually being presented (because we conservatively lower bound peptides against 11 different binding prediction models). This work lays the foundation for a new automated epitope based vaccine design pipeline which can design a vaccine for any pathogen (or cancer) provided.
Read the paper here: https://www.biorxiv.org/content/10.1101/2020.05.16.088989v1
Paper summary thread 1/n.

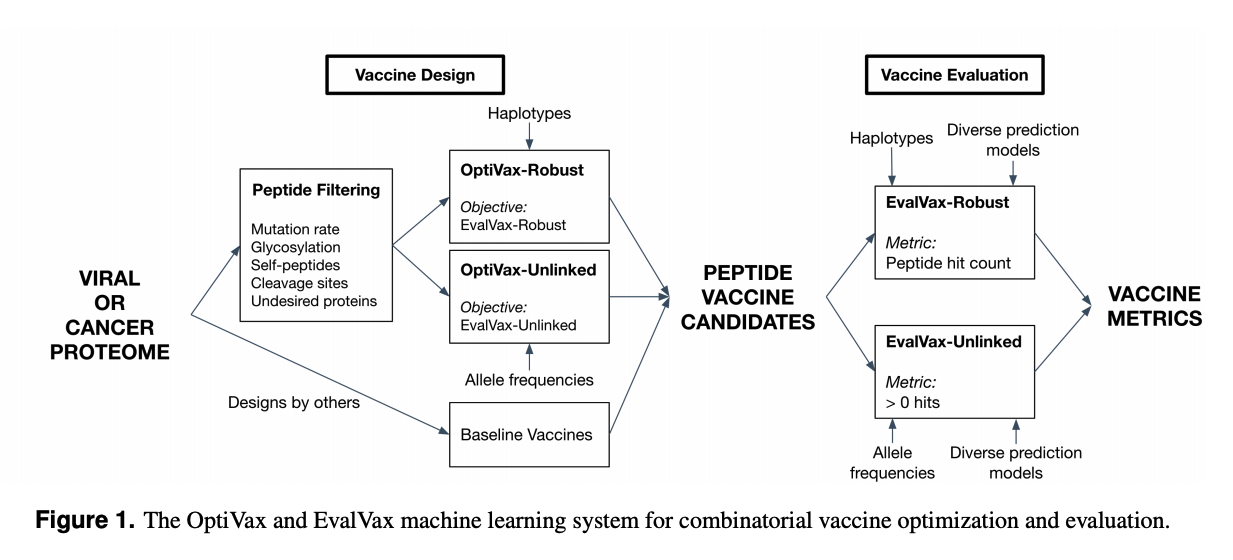
We introduce a novel population coverage objective function that calculates the probability of at least n peptides from the vaccine being presented by an individual. We develop a variant of the objective (EvalVax-Robust) that also accounts for the strong linkage disequilibrium that exists across HLA types.
In selecting peptides, we avoid peptides where SARS-CoV-2 has significantly mutated, crosses a cleavage site, is predicted to be glycosylated, or matches a self peptide. We also use 11 different epitope binding prediction algorithms in total to conservatively select peptides predicted to strongly bind (<50nM) across all of them.
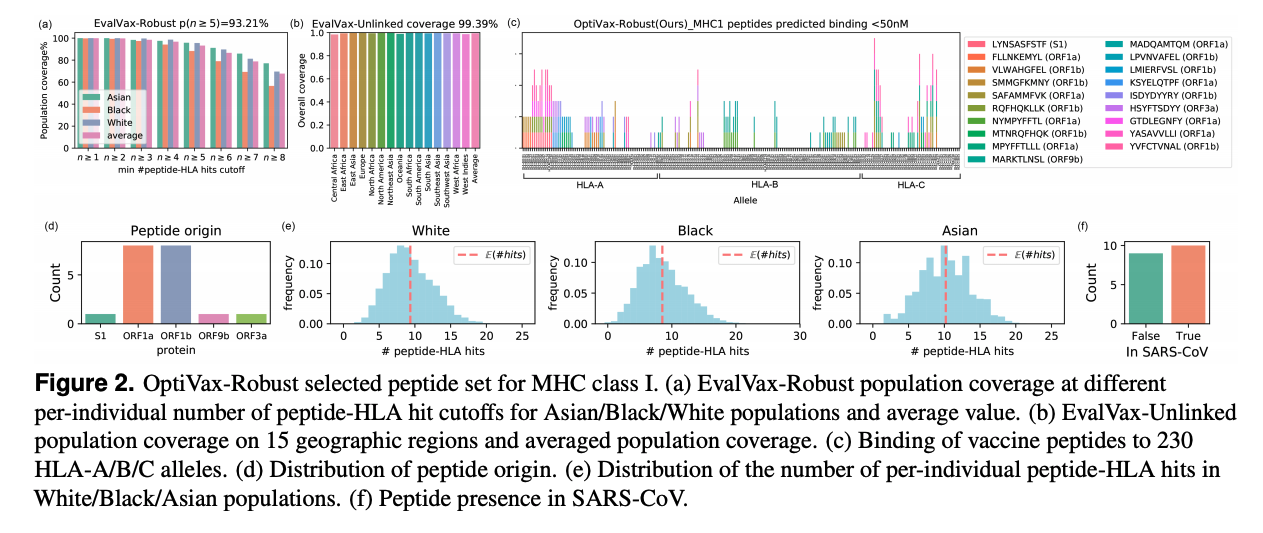
Here is one of our designs for MHC-1 (for CD8-T cells) that accounts for linkage disequilibrium in HLA frequencies present across different races. It consists of 19 peptides and is predicted to have at least 5 peptides bind in 93.2% of the global population (top left). Also note the diversity of peptide origins across the SARS-CoV-2 proteome (bottom left) and the potential for half of these peptides to be broadly neutralizing because of their presence in SARS-CoV (bottom right).
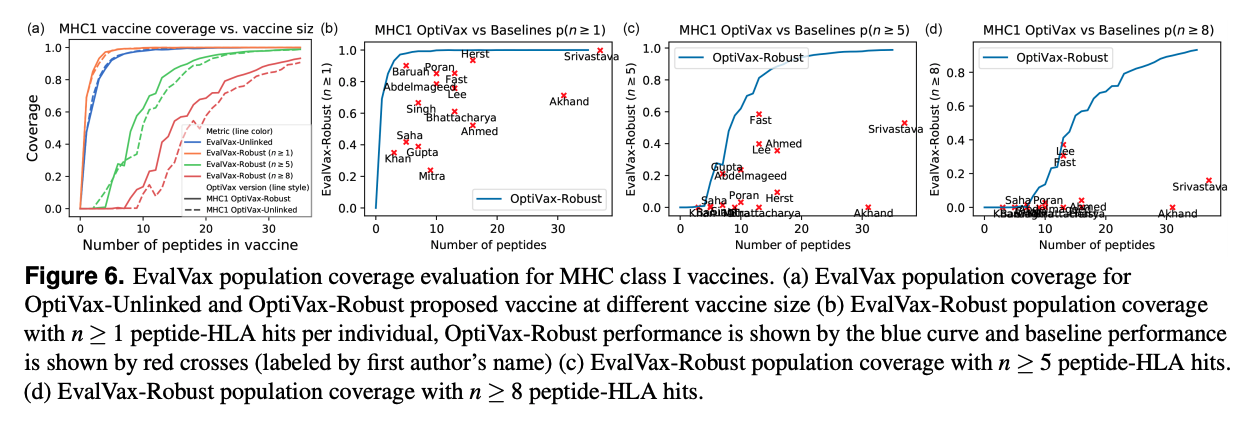
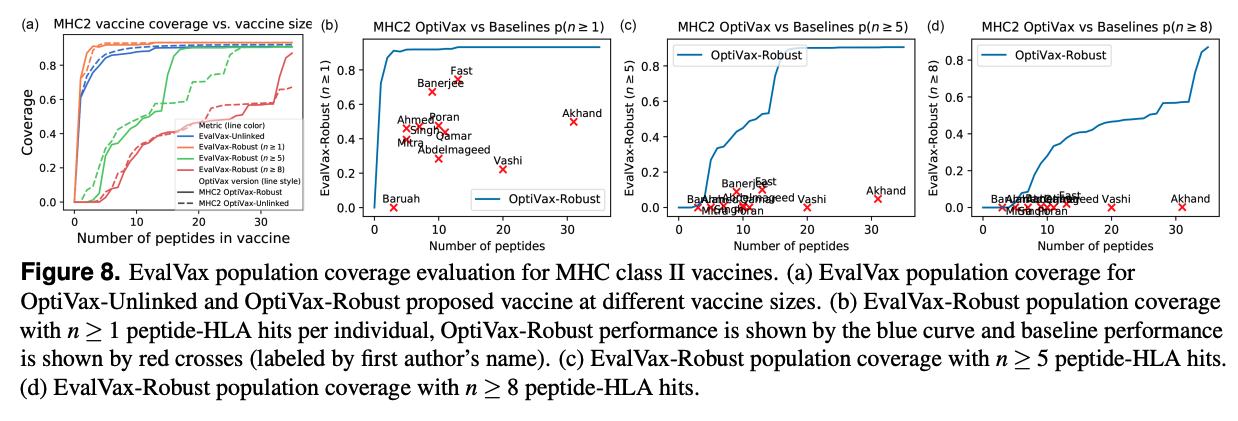
Using this pipeline, our SARS-CoV-2 vaccine is shown to outperform every other epitope based SARS-CoV-2 vaccine published to date. As shown below for MHC 1 and 2, independent of the number of peptides in the vaccine, we have a higher predicted population coverage.
We will hopefully test our vaccine in animal models. How does this work help our current pandemic situation? In addition to turning it into a future automated pipeline to combat the next pandemic, our vaccine leverages a diverse set of peptides across the viral proteome that have not mutated to date and thus presents a nice addition to our current vaccine portfolio because many vaccines under development are Spike protein only (there is no evidence to date that having only Spike protein won’t work great but these remain early days).
Thanks to the Gifford lab and their collaborators for being such a pleasure to work with and giving me the opportunity to contribute to this important research :)
Note that this is a preprint. If you read the paper do send feedback. Making a COVID vaccine is too important for mistakes to be made.
——> Paper: https://www.biorxiv.org/content/10.1101/2020.05.16.088989v1